.png)
Demystifying IDMP – All you need to know to get started!
October 11th, 2022 WRITTEN BY Aditi Acharya. Sr. Manager, Client Success Tags: Data, data management, IDMP, Life Sciences

Written By Aditi Acharya. Sr. Manager, Client Success
What is IDMP?
If you work in the Life Sciences industry, the term “IDMP” would be familiar to you. IDMP stands for Identification of Medicinal Products which is a set of standards used to globally standardize data and structures to define and uniquely identify medicinal products. IDMP comprises of a set of five standards developed by ISO, or the International Organization for Standardization.
The five ISO IDMP standards, all of which deal with unique identification and exchange of information for medicinal products, are:
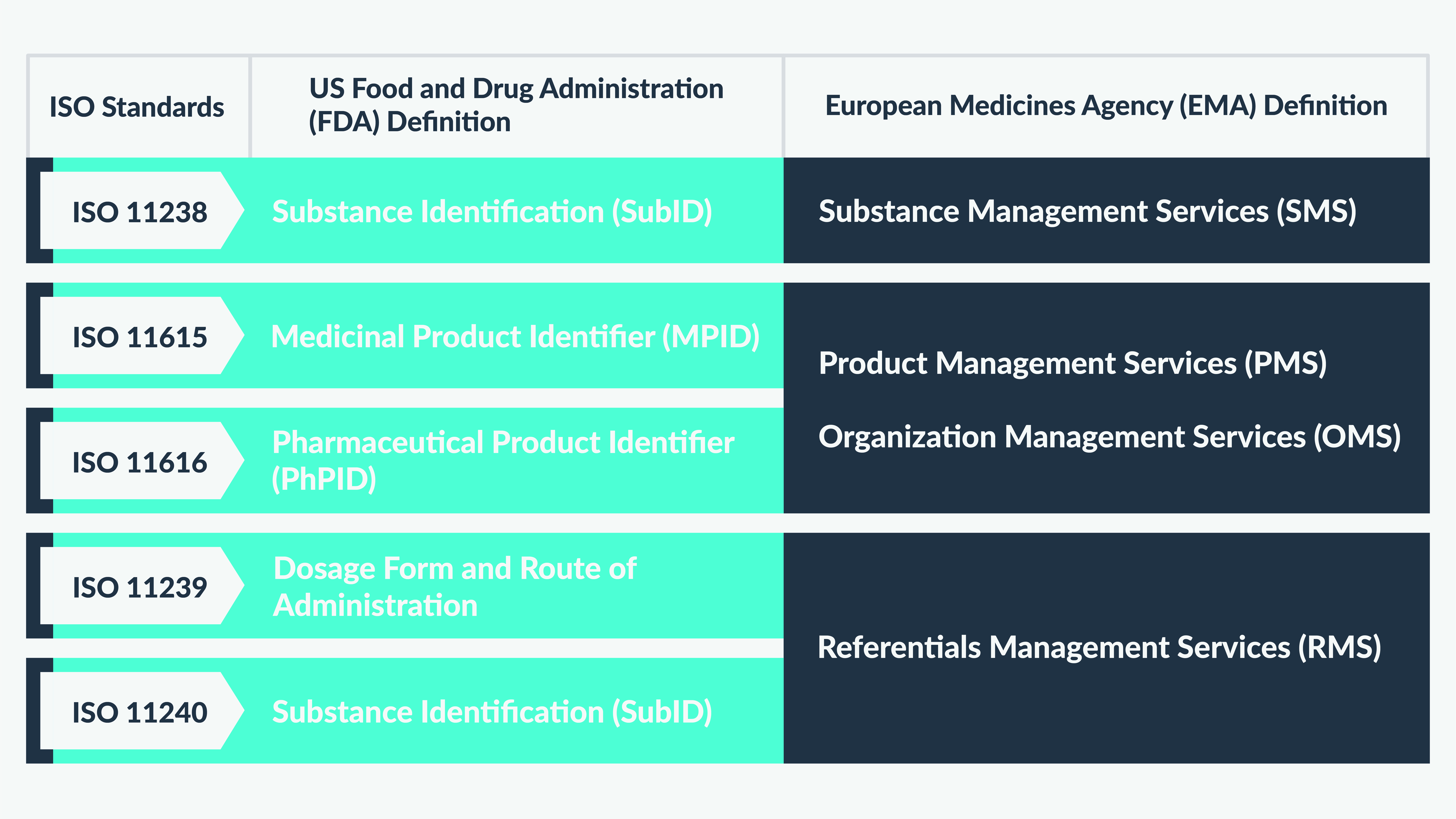
- ISO 11615: Standards relating to Medicinal Product
- ISO 11616: Standards relating to Pharmaceutical Product
- ISO 11238: Standards relating to Substances.
- ISO 11239: Standards relating to pharmaceutical dose forms, units of presentation, routes of administration, and packaging items related to medicinal products.
- ISO 11240: Standards relating to units of measurement.
The 5 ISO standards are illustrated and described in Figure 1:
 Why IDMP?
Why IDMP?
The competitive, dynamic, and highly governed nature of the Life Sciences industry requires a continuous flow and exchange of data between regulatory authorities, pharmaceutical companies, and manufacturers, among other stakeholders. While there has always been a need for efficiently managing submissions and adverse events reporting, there lacked a single mechanism to reliably exchange accurate information between stakeholders. To address this need in the context of pharmacovigilance and improving adverse event reporting, IDMP was developed.
ISO IDMP provides a standard way for defining and storing medicinal product information, which will enable efficient reporting, tracking, faster decision-making, and response during adverse event reporting. At present IDMP focuses on standardization of data with a future goal of improving overall pharmacovigilance[1].
Currently, most of the medicinal product information is spread across fragmented systems within pharmaceutical organizations. Some of the systems are legacy systems and hold data in an unstructured format such as documents, pdfs, excel workbooks, and email messages. Standardizing this data in the IDMP format supports the regulatory submission processes within an organization as it not only maintains uniformity in managing data assets within an organization but also in exchanging data between the regulators (such as the EMA and the FDA) and Life Sciences organizations. The Medicinal Product data previously submitted to regulatory authorities can be re-used when submitting variations to authorized medicinal products. In case of additional requests from regulatory authorities for a submission or a query, the information will be readily accessible. For an Investigational Medicinal Product, as the clinical trial progresses, data generated through different business processes can be submitted periodically and standardization in IDMP format will help maintain transparency.
What has happened so far?
The European Medicines Agency (EMA), a regulatory agency of the European Union (EU) is pioneering the IDMP journey and has organized the implementation using its SPOR services. SPOR is a set of four data management services, namely:
- Substance Management Service (SMS)
- Product Management Service (PMS)
- Organization Management Service (OMS)
- Referential Management Service (RMS)
SPOR covers multiple master data domains of the medicinal products definition. IDMP is being implemented by EMA in phases with different timelines. RMS and OMS services were launched in 2017 and are currently used in submissions that need to be made to the EMA and other regulatory authorities within Europe. As per timelines published by EMA, the next in line is the implementation of the Product Management Service (PMS), expected to go live in Q2 2023. This means that all submissions for medicinal products will need to be made to the PMS service using web-based Digital Application Dataset Integration (DADI) forms (Latest Implementation Guidance IG v2.1 published here). Regulatory organizations, just like the EMA, will be holding the beacon for guidance on IDMP compliance for the industry.
The United States Food and Drug Administration (FDA) has not yet mandated the use of IDMP standards for the submission of data to the FDA. Nevertheless, preparation for EMAs SPOR services will also ensure readiness for FDAs’ future requirements.
Figure 2 is an illustrative depiction of the different approaches undertaken by the US FDA and EMA for defining IDMP ISO standards.
What next?
As IDMP timelines for EMA draw near, Life Sciences organizations are embarking on a digital transformation journey. At a strategy level, an organization can lay down a roadmap for IDMP and align it with its long-term goals to build Enterprise level Data Assets. IDMP compliance is to be viewed as an opportunity to break organizational data silos, improve overall organizational data quality and governance, and enhance operational efficiencies.
Organizations will have to be agile to implement IDMP. Life Sciences organizations and regulators will have to work in collaboration to fully leverage the adaptive, scalable, and nimble technologies available in the market, to achieve 100 percent IDMP compliance.
Did you know about our IDMP solution?
Fresh Gravity has built an MDM-driven solution to address IDMP Compliance needs. Read more about Fresh Gravity’s approach to this here.
For a demo of Fresh Gravity’s solution, or more information and questions, please write to neha.inamdar@freshgravity.com.
For more detailed information about this solution, please refer to the datasheet here.
[1] Pharmacovigilance is the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other medicine/vaccine-related problem. [Source:who.int].
Share this




